
Research outline
Research in the life sciences field has undergone a paradigm shift to a data-driven style since the human genome sequencing completion, because of the large amount of available genomic information. Recently developed massive parallel sequencing technologies have helped accelerate its momentum globally; however, the response of Japanese universities and other research institutions to this shift remains lagging. There is an urgent need for infrastructure development, in order to create a research community and scientific bases to make society accept the importance of massive data management and information/knowledge extraction that would play a major role in the future development of information and genome science.
Therefore, in this project, the latest genome-based technology is to be utilized to generate and collect systematically genomic and genetic information at a large-scale, in addition to pluralistic phenotype data in order to develop a method for the statistical analysis of various types of information. Also, a generic description of genomic function and genetic network will be prepared by integrating all obtained data, and a statistical method developed to describe the genetic correlation structures. This method is to be refined by applying to a large volume of genomic and phenotypic information obtained from a specific model organism. Through this, we aim to understand the biodiversity in a system resulting from the high-order association of a number of genetic factors.
The project includes the exchange of information between groups investigating three subthemes, in order to develop a new method for researching life phenomena. This will be done with an aim to propose a novel interpretation and principle, unique to data centric science.
(Project Director: Nori Kurata - National Institute of Genetics).
Purpose of the project
The major purpose of this project is to develop a statistical method for the modeling of multidimensional diversity of biological phenotypes, as well as a method for the analysis of massive genome sequence and gene expression data. A novel method could be developed by integrating these two methods, in order to visualize complex genetic correlation structures. This method is to be ultimately applied to a model organism, in order to extract the genomic function and network.
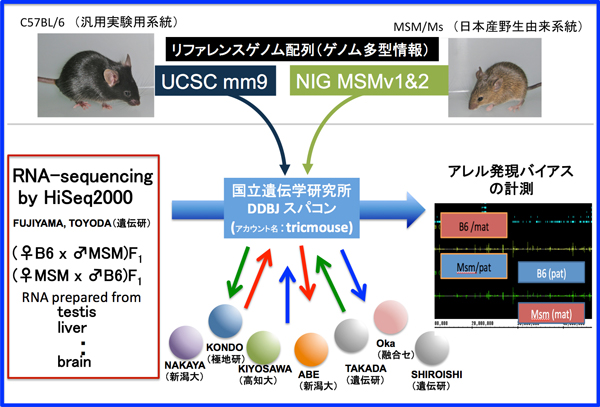
Project Promotion System
Studies analyzing large volumes of data for genetic experiments and genomic information via various informatics or statistical processing methods, as well as those deciphering, reconstructing, and utilizing a massive amount of genomic information, have seen increasing popularity in the West. These studies are essential for solving the problems in food, environmental, and medical sciences; therefore, a comprehensive research system must be quickly established.
In this project, therefore, massive and pluralistic genetic information is to be comprehensively analyzed through genetic, informatics, and statistical methods in order to establish a “genetic function system science”. In addition, a study will be conducted to understand the complex principles of life and genetic phenomena as a system. Initially, exhaustive data for multidimensional and diverse genetic factors, such as massive genome sequence polymorphism information, gene expression variation information, phenotype variation, temporal changes of those characteristics, etc. will be obtained using the genetic resources owned by the National Institute of Genetics. Finally, the information processing technology of the National Institute of Informatics and statistical modeling technology of the Institute of Statistical Mathematics is to be utilized to determine the genomic function and genetic network.
Introduction of subthemes
1. Large-scale production of genome-related information through next-generation sequencing, and the development of a method to analyze massive information
The latest genome technology will be used to systematically and exhaustively produce massive-scale genomic information, complex genome system-derived and gene-related data. A fusion research is to be conducted utilizing these data with the goal of comprehensively understanding the life system principle. To this effect, this subtheme incorporated multiple topics. In addition, the research will be performed under collaboration with the groups investigating subthemes 2 and 3; this research would also coordinate with other integrated projects focusing on massive-scale sequencing
(Principal Investigator: Asao Fujiyama - National Institute of Genetics/National Institute of Informatics).
- Massive-scale genomics: A next-generation large-scale genome sequence analysis pipeline will be constructed in order to execute the production and processing of a large volume of genomic data; in addition, we will produce genome sequence data and gene expression data from genetically diverse groups, in collaboration with the investigators of subthemes 2 and 3. In the course of developing diverse methods for the analysis of these data, and stratifying the data jointly with the results of subthemes 2 and 3, we aim to visualize the obtained data, and publish the results to the research community via the Internet.
- Advanced genomics in the functional genomic region: We aim to implement a large-scale genome analysis to elucidate basic biological phenomena, such as genome replication, gene expression regulation, developmental regulation of individual organism, epigenetic control, etc. In addition, important genomic functional regions, not analyzed by conventional genomic research because of their complex structure, such as the centromere and telomere regions are analyzed. We also aim to promote the interdisciplinary development of a method for the comprehensive analysis of this data.
2. Development and optimization of a statistical method for the visualization of genetic correlation structures, through the integration of a large volume of genome-related and pluralistic biological phenotype diversity data.
The main aim of this set of studies is to develop correlation analysis, regression analysis, permutation test, robust inference, impact analysis, graphical model, multiplicity adjustment, and other probability and statistical models relative to linkage analysis, quantitative trait loci (QTL) analysis, eQTL analysis, genetic network structure identification, and genetic diversity analysis. Specifically, this includes the following topics:
- Equivalence test of variance
- Development of a multiplicity adjustment method that takes into consideration the correlation of genome data
- Development of a graphical model for the genetic information and expression data
- Preparation of methodology for diversity analysis of human genome sequence, and establishment of an analytical method using QTL regression analysis in proximity.
- A method in which dimensional compression of high-dimensional explanatory variables is performed in accordance with the expected purposes.
(Principal Investigator: Satoshi Kuriki - The Institute of Statistical Mathematics).
3. Extraction of genetic networks and genomic functions by applying informatics and statistical methods on a large amount of pluralistic data.
Mice, zebrafish, drosophila, and rice are models that have been uniquely developed and collected by the National Institute of Genetics. These are valuable research models containing a host of genomic and phenotypic information. Informatics and statistical methods established by the investigators of subthemes 1 and 2 will be applied to a large amount of model organism genomic and phenotypic data. With the further evaluation and improvement of the utilized methods, we aim to expand the foundation of “genetic function system science”, wherein the generic extraction of genetic networks and genomic functions would be possible.
- Mouse: Includes the quantification of complex traits using wild strain and consomic (chromosome substitution strains) mouse models, data extraction, and development of a statistical analysis method. Genome diversity data and correlation analysis.
- Rice: Includes the analysis of intersystem genome structure of wild rice and lineage population structure, analysis of gene expression variation, method for selection of phenotypes, improvement of the association analysis method, and a correlation analysis between genome diversity and functional/phenotypic variation.
- Zebrafish: Analyses include identification of phenotypes using various transgenic fish strains, establishing a method to convert to data, and the identification of genotype correlation.
- Drosophila: Analyses to be performed include the development of a method to identify variations in the wings using multiple genetically-modified strains, and analysis of the correlation with modified genes.
(Principal Investigator: Nori Kurata - National Institute of Genetics)

Research View 029
The NEXT STEP of Genome X Statistics
[Genetic Function Systems] Satoshi Kuriki (Professor, The Institute of Statistical Mathematics), Hironori Fujisawa (Professor, The Institute of Statistical Mathematics)

Research View 028
Repatriation of the “Bean-Spotted” Fancy Mouse
[Genetic Function Systems] Toshihiko Shiroishi (National Institute of Genetics, Vice Director & Professor)

Research View 009
What is the Mechanism by Which a "Species" is Formed?
[Genetic Function Systems] Ayako Oka (Project researcher, Transdisciplinary Research Integration Center)

2014-04-18 Press Release
A mechanism of genetic regulation evolving in different directions leads to new species!
[Genetic Function Systems] Ayako Oka (Project researcher, Transdisciplinary Research Integration Center), etc.

Research View 002
Whole genome sequence tells a story: where does cultivated rice come from?
[Genetic Function Systems] Nori Kurata (Professor, National Institute of Genetics)